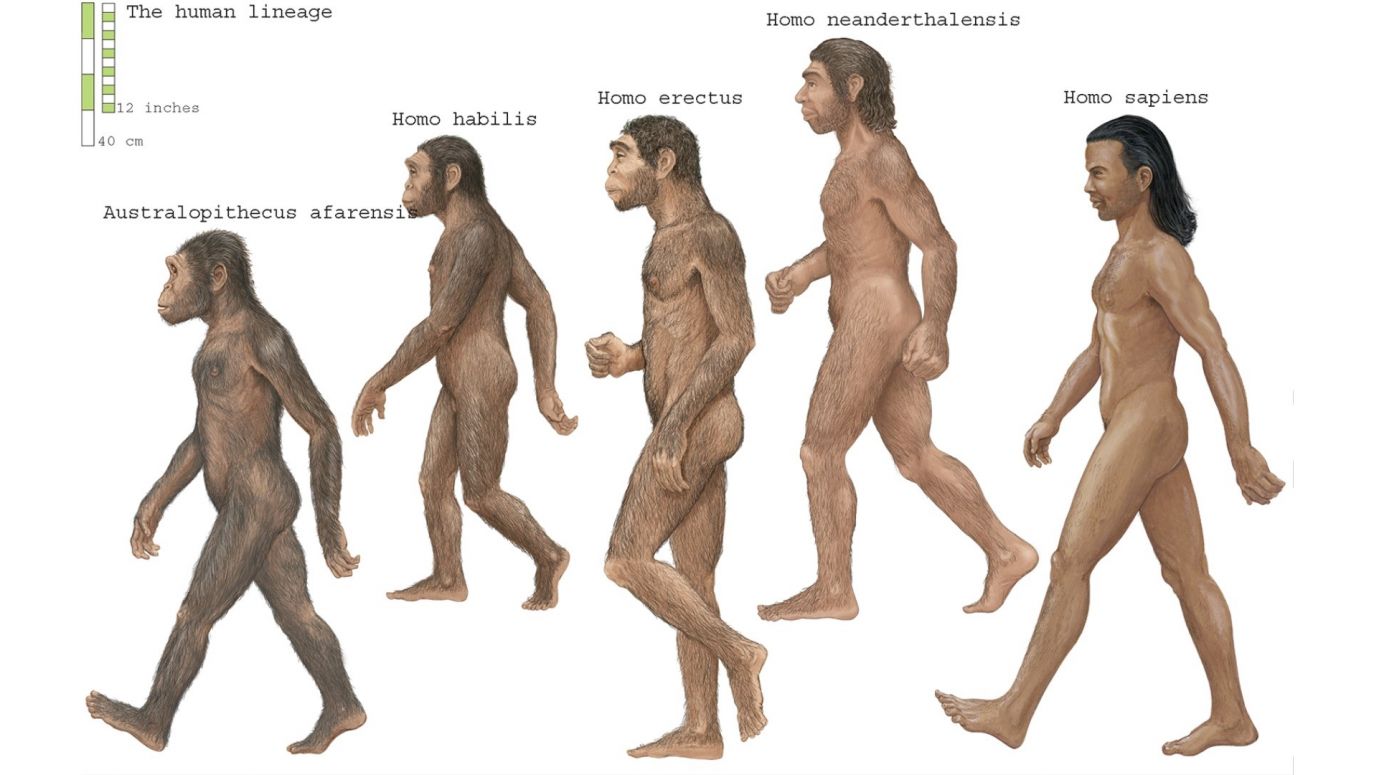
Ashton i Stringer uznają: „Rozważenie, w jaki sposób wywnioskowane wąskie gardło mogło wpłynąć na ewolucję człowieka, nieuchronnie prowadzi do debaty na temat natury ostatniego wspólnego przodka
H. sapiens, neandertalczyków i denisowian oraz tego, kiedy i gdzie żył ten przodek. Nie jest jeszcze jasne, czy ostatni wspólny przodek żył w Europie, Azji czy Afryce. […] Co więcej, niektóre modele genetyczne głębokiego pochodzenia
H. sapiens i neandertalczyków sugerują, że koncepcja ostatniego wspólnego przodka w czasie i przestrzeni może być iluzoryczna. Szacunki wykorzystujące dane genomiczne
H. sapiens, neandertalczyków i denisowian kalibrują ostatniego wspólnego przodka na okres od około 500 tys. do 700 tys. lat temu, co wiązałoby wywnioskowane wąskie gardło z tą populacją przodków, niezależnie od tego, gdzie mieszkała […]”.
To nie jedyne konsekwencje. „Wąskie gardło prowadzi nie tylko do spadku różnorodności genetycznej populacji, ale też do losowej akumulacji wariantów niekorzystnych, z którymi w liczniejszej populacji poradziłby sobie dobór” – wyjaśnia prof. Golik. „Trudno powiedzieć, jakie konkretnie zmiany związane były z tym okresem redukcji liczebności populacji. Na pewno przyczynił się on do szybszych zmian genetycznych u przodków ludzi. Mógł też odegrać znaczącą rolę w ich rozdziale na różne gatunki (neandertalczyka,
Homo sapiens i denisowian). Przyczynił się też do tego, że człowiek współczesny, mimo ogromnej obecnie liczebności populacji, jest gatunkiem o stosunkowo małym zróżnicowaniu genetycznym. Choć dużą rolę odegrały tu też późniejsze wąskie gardła”.
Musimy sobie bowiem zdawać sprawę, że takie populacyjne wąskie gardła przydarzały się nam częściej. Jeśli nie globalnie, to co najmniej lokalnie. Sama koncepcja „out of Africa” zakłada, że kilkukrotnie migrowały z Afryki niewielkie populacje naszego gatunku i jego przodków. A zatem cała dzisiejsza populacja euroazjatycka pochodzi tylko z niewielkiego ułamka przodków współczesnej rdzennej populacji Afryki. Jesteśmy zatem – natywni Euroazjaci – od natywnych Afrykańczyków znacznie mniej bioróżnorodni. Kolejne wąskie gardło to migracja z Eurazji na kontynent amerykański. Wszystko zaczęło się od grupy mieszkańców Syberii, nie większej niż 250 ludzi, wędrujących niekoniecznie dawniej niż 23 tys. lat temu. Co więcej, gdy ich potomkowie osiągnęli w pamiętnym roku 1492 liczbę 60,5 mln, to następnych sto lat przeżył jedynie co dziesiąty z nich. Uszczuplenie puli genowej o 90 procent – a z takim mieliśmy do czynienia w kolumbijskiej Ameryce, takim samym jak według chińskich analityków 900 tys. lat temu wśród naszych przodków homininów – to hekatomba.
„Aby przetrwać, ta populacja na granicy wymarcia zajmowała bardzo zlokalizowany obszar o dobrej spójności społecznej” – piszą Ashton i Stringer. Jeszcze większym zaskoczeniem według nich jest fakt, że ta mała grupa trwała, nie powiększając się, aż 117 tys. lat. „Jeśli to prawda, można sobie wyobrazić, że wymagałoby to stabilnego środowiska z wystarczającymi zasobami i niewielkim obciążeniem systemu”.
 ODWIEDŹ I POLUB NAS
ODWIEDŹ I POLUB NAS

Na uzyskany przez Chińczyków model można spojrzeć krytycznie. Prof. Golik przypomniał, że „badania różnorodności genetycznej człowieka wciąż trwają i nie ma pewności, czy kolejne wyniki nie zmuszą nas do rewizji tych założeń. Do niedawna większość badanych ludzkich genomów pochodziła spoza Afryki. W omawianej pracy próbki pochodziły z 10 populacji afrykańskich i 40 spoza Afryki. Tymczasem wiemy, że mieszkańcy Afryki stanowią grupę o wiele bardziej różnorodną, niż cała reszta świata razem wzięta. Już w 2020 roku opublikowano pracę obejmującą próbki z 50 tamtejszych populacji. A 50 to mały wycinek, bo różnych grup etnolingwistycznych w Afryce są jakieś 2 tysiące. Być może, gdy lepiej poznamy tę różnorodność, będziemy musieli zmienić modele prehistorii naszego gatunku. Zauważyć też trzeba, że w badaniu z Szanghaju żadnych ciekawych mutacji nie przypisano konkretnie do tego okresu wąskiego gardła”.
Pozostaje zagadką, co się stało ok. 800 tys. lat temu, że populacja naszych przodków zaczęła rosnąć – i to dość gwałtownie. Zanim to nastąpiło, straciła 2/3 całej swej różnorodności genetycznej. Czy owo wąskie gardło było naprawdę globalne? I jak się nam dziś żyje bez tych wszystkich utraconych wariantów genetycznych? Czy tak dramatyczna selekcja oszlifowała nas jak diament (stąd np. późniejszy gwałtowny rozrost puszek mózgowych naszych przodków)? Pytań raczej przybyło niż ubyło, choć zniknęło 90 proc. potencjalnych obiektów badawczych, bo nie zostawiły one śladu w naszych współczesnych (przebadanych) genomach.
– Magdalena Kawalec-Segond
TYGODNIK TVP, ul. Woronicza 17, 00-999 Warszawa. Redakcja i autorzy
Źródła:
https://www.science.org/doi/10.1126/science.abq7487
https://www.science.org/doi/10.1126/science.adj9484
https://www.nature.com/articles/d41586-023-02712-4
https://www.nature.com/articles/s41586-020-2859-7